近日,我院范海龙副教授团队在力响应高分子材料领域取得重要进展:他们受天然蛋白质中局部序列组织与动态非共价相互作用协同调控力学响应的启发,构建了具有统计富集相邻阳离子–芳香基序的疏水聚电解质。该聚合物的高浓度水溶液可在剪切作用下由黏性液体转变为瞬态凝胶,并在撤去剪切后恢复流动;即使在盐溶液中形成凝聚结构,这种可逆剪切诱导凝胶化行为仍可保持。相关成果发表于《Macromolecules》。公海jcjc5500陈尹安为论文第一作者,范海龙副教授为通讯作者,公海jcjc5500为第一完成单位。

阳离子-π相互作用广泛参与蛋白质折叠、分子识别、相分离及湿态黏附等生命过程,其通常依赖阳离子基团与芳香基团在局部空间上的紧密邻近排布。但在水相环境中,芳香基团易发生疏水聚集,水合作用也会削弱短程的阳离子-π相互作用。同时,传统共聚方法通常只能调控单体的总体组成,难以控制单体在链上的序列分布。
针对这一难题,研究团队选取反应活性相近的阳离子单体 ATAC 与芳香单体 PEA 进行理想恒比共聚,制备得到聚合物 P(ATAC-stat-PEA)。该合成策略在聚合物链上实现相邻阳离子-芳香基序的统计富集。链上相邻的阳离子单元可充当间隔基团,抑制芳香基团在水中发生直接疏水聚集,同时保留了形成阳离子-π相互作用所需的局部空间构型。
流变测试结果显示,P(ATAC-stat-PEA) 高浓度水溶液在持续剪切作用下黏度随时间显著增加,原本可自由流动的溶液逐渐转变为具有类固体力学特征的瞬态凝胶;撤去剪切后,凝胶网络逐步松弛,样品恢复流动状态。循环流变测试进一步证实,这种 “液体-凝胶”的转变具备良好的可逆性与可重复性。相比之下,将芳香单体替换为非芳香单体制备的对照聚合物仅表现出常规的剪切变稀行为,表明芳香基团及其与阳离子单元构成的局部基序是诱导该现象的关键因素。
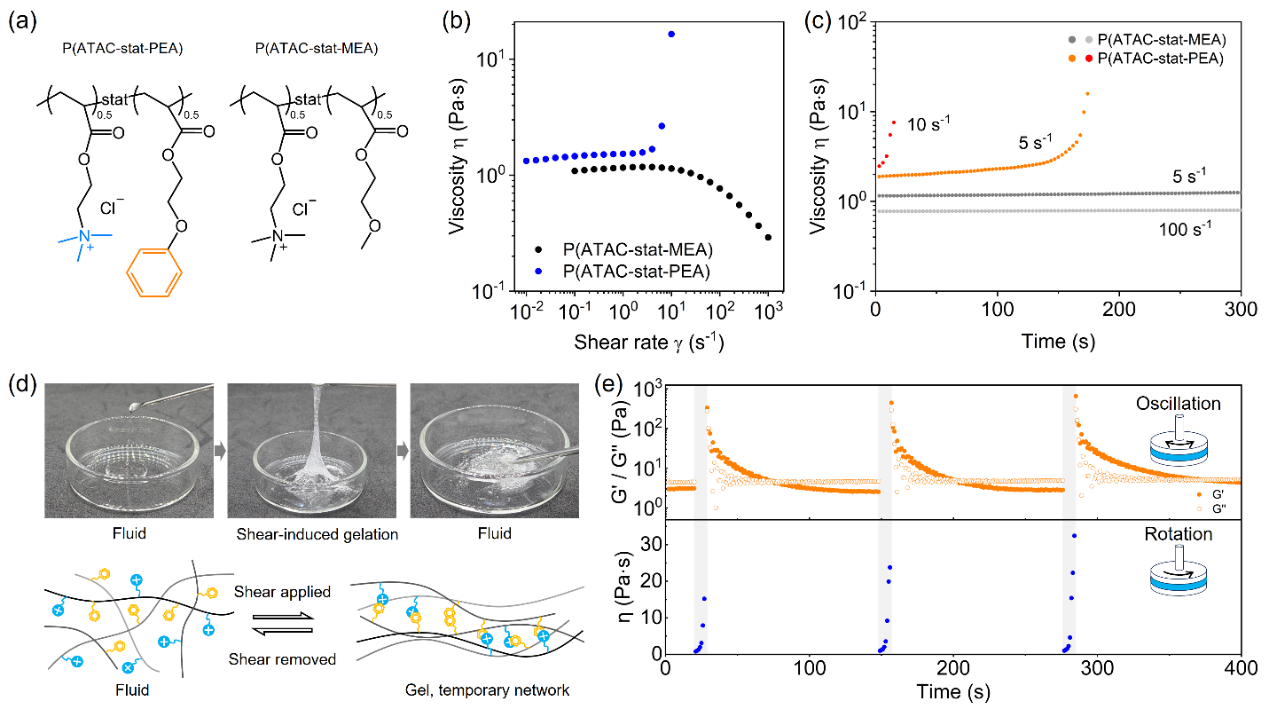
图1. P(ATAC-stat-PEA)水溶液的可逆剪切诱导瞬态凝胶化行为
研究进一步发现,该剪切诱导凝胶化行为受聚合物浓度与剪切速率的共同调控。随着体系浓度升高,聚合物链间距减小,链重叠/缠结程度增大,体系更易在剪切作用下形成贯穿性的网络结构。小角X射线散射(SAXS)与二维核磁共振结果表明,静置的聚合物水溶液中已存在浓度依赖的链间关联和短程阳离子–芳香空间邻近,为剪切过程中链间动态连接的形成提供了结构基础。

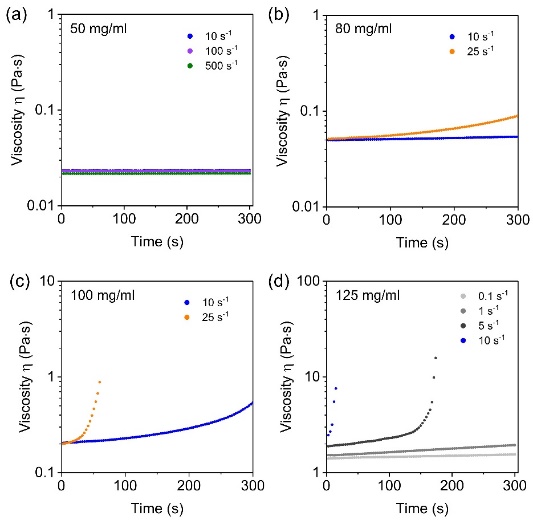
图2. P(ATAC-stat-PEA)水溶液中链缠结状态的浓度依赖性及剪切诱导凝胶化动力学
向体系中加入盐后,静电屏蔽效应会驱使聚合物形成更为致密的凝聚结构,但可逆剪切诱导凝胶化的特性依然得以保留,且表现出显著的离子种类依赖性。相同实验条件下,凝胶化速率遵循LiCl>NaCl>KCl的规律;小角X射线散射结果显示,盐溶液中形成的棒状聚集体半径也随LiCl、NaCl、KCl的顺序逐渐减小。
这一变化规律与水相中碱金属离子竞争阳离子-π作用的趋势一致。Li+水合作用更强,对芳香位点的竞争能力较弱,可保留更多聚合物阳离子与芳香基团之间的链间连接;K+对芳香位点的竞争能力更强,会更显著地扰动聚合物内部的阳离子-芳香关联。上述结果表明阳离子-π相互作用是驱动该体系产生剪切凝胶化的主要驱动力,疏水作用协同参与。
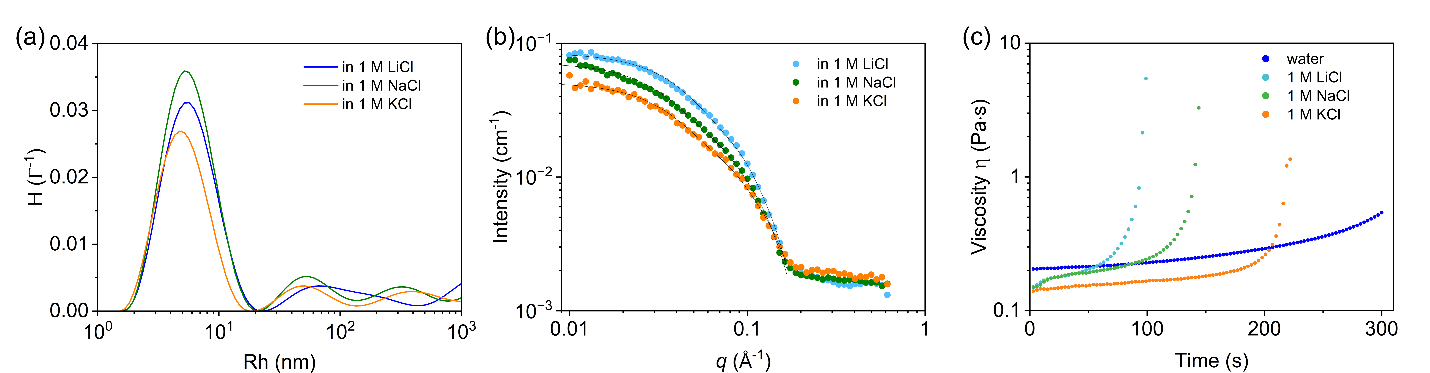
图3. 盐环境中P(ATAC-stat-PEA)的离子调控组装行为及剪切诱导凝胶化特性
该研究表明,合成高分子的力响应行为不仅可通过引入特定功能基团进行调控,还能通过控制功能单元在聚合物链上的局部统计排布实现编码。与复杂的精准序列合成方法相比,本研究仅通过常规自由基共聚即可构建具备类蛋白质局部基序的聚合物,为设计兼具可逆力响应、自适应流变与动态结构重构能力的软材料提供了全新的设计思路。
文献链接:
https://doi.org/10.1021/acs.macromol.6c00991